
Computational Design of Functional Materials Using Density Functional Theory for Energy Applications
DOI:
https://doi.org/10.53762/grjnst.04.02.11Keywords:
Adsorption Energy, Density Functional Theory, Energy Materials, Graphene, Perovskite, SemiconductorAbstract
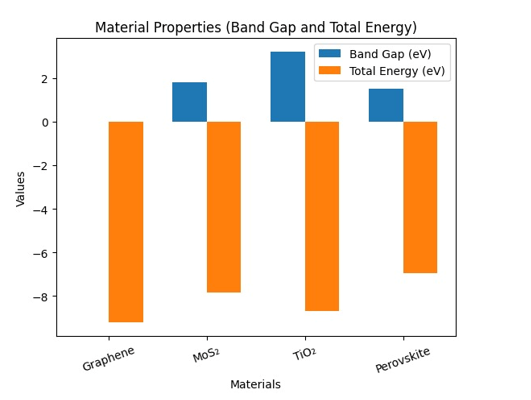
This study investigated the computational design of functional materials for energy applications using Density Functional Theory (DFT). The primary objective was to analyze the structural, electronic, and catalytic properties of selected materials to evaluate their suitability for energy storage and conversion systems. A quantitative computational methodology was adopted, where materials such as graphene, molybdenum disulfide (MoS₂), titanium dioxide (TiO₂), and perovskites were analyzed using DFT-based simulations. Key parameters including band gap energy, total energy, density of states, and adsorption energy were calculated. The results revealed that graphene exhibited a band gap of 0.00 eV, indicating high electrical conductivity, while MoS₂ and perovskites showed moderate band gaps of 1.80 eV and 1.50 eV, respectively, making them suitable for photovoltaic applications. TiO₂ demonstrated a higher band gap of 3.20 eV, suggesting its suitability for photocatalytic processes. Adsorption energy analysis showed that MoS₂ (−0.85 eV) and perovskites (−0.65 eV) had optimal interaction strengths for catalytic efficiency, whereas graphene exhibited weak adsorption (−0.20 eV). The findings highlighted that DFT-based approaches significantly enhanced the efficiency of material design by reducing experimental efforts and enabling accurate prediction of properties. The study provided practical implications for developing advanced energy materials and emphasized the importance of computational techniques in achieving sustainable energy solutions.
Downloads
Published
Issue
Section
License
Copyright (c) 2026 Irtaza Bashir Raja, Hamid Iqbal, Muhammad Muneeb Khan, Sheraz Ahmad (Author)

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.




